 |
 .
. .
.  .
.
Universidade Federal de Minas Gerais.
Belo Horizonte, Minas Gerais, 2000.
Se você tiver alguma sugestão ou comentários,
por favor envie-os para: Laboratório de Apoptose/ Departamento
de Patologia Geral/ Instituto de Ciências Biológicas da
Universidade Federal de Minas Gerais, Belo Horizonte, MG 31270 010,
Fone: (0xx31) 499 28 87/ 499 28 81 / 9114 78 41
FAX: (0xx31) 499 28 78,
Web
http://www.icb.ufmg.br/~pat/apopt.htm
.e-mail:anilton@icb.ufmg.br
|
 NEOPLASIAS
NEOPLASIAS
Oncogênese
( bases, fases, teorias, mecanismos gerais ); condições
pré-malignas; graduação e estadiamento;
"Os tumores são excrescências
atípicas provenientes dos germes embrionários".
JULIUS CONHEIN, 1882.
"Cancer is a complex, multistage, multimechanism
phenomenon."
J. MICHAEL BISHOP
- ONCOGÊNESE:
- Teorias
da oncogênese - Histórico:
- Da irritação crônica (VIRCHOW);
- Do desvio embrionário (CONHEIM);
- Da diferenciação defeituosa pós
embrionária (RIBERT)
- Do desvio metabólico (WARBURG, 1925/25): "O
crescimento descontrolado dos oncócitos se relaciona com desvio
metabólico expresso pela glicólise aeróbica".
- Da uniformização bioquímica
dos oncócitos (GREENSTEIN, 1955)
- Da deleção de repressores (MILLER,
1945): Perda ou alteração permanente de fatores protéicos
capazes de controlar o crescimento celular.
- Da mutação
(de células somáticas, por cancerígenos):
- A proliferação das células
normais é regulada de modo inibitório pelas proteínas
codificadas pela expressão dos Genes Supressores de Tumores
e de modo estimulatório pelas proteínas codificadas pela
expressão dos proto-oncogenes. Além dos oncogenes
e dos genes supressores de tumores (anti-oncogene), os genes que condicionam
a morte celular programada (apoptose), a mobilidade celular e a produção
de Fatores do Crescimento estão também envolvidos no processo
de oncogênese.
- As neoplasias decorrem da perda ou inativação
(por mutação ou por herança de uma cópia
defeituosa) de ambas as cópias dos genes supressores de tumores
(mutação recessiva), freqüentemente associado
à hiperativação (por mutações, transformando
o proto-oncogene em oncogene, por amplificação do número
de cópias do gene, por translocação cromossômica;
ou por inserção de um retrovirus oncogênico, integrando-se
próximo a um proto-oncogene no genoma do hospedeiro) de uma
das 2 cópias dos proto-oncogenes(mutação dominante).
Os genes dos Fatores do Crescimento, da apoptose e dos genes do
mobilidade celular com frequencia tambem estão envolvidos.
Estudos moleculares revelam que no câncer há
sempre um coleção complexa e heterogênea de lesões
genéticas ("Multiple Hit Hipothesis").
Oncogenes:
normalmente encontrados nas células (proto-oncogene), mas que quando
afetados por mutações (dominantes) ou translocações
codificam proteínas superexpressados que desviarão a cascada de
eventos que controlam a proliferação celular, no sentido de uma
proliferação incontrolada (levando às neoplasias).
Quando uma célula produz o seu próprio fator de crescimento, ela
adquire AUTONOMIA.
|
Oncogene
|
=
|
Acelerador |
|
Mutações nos oncogenes
|
=
|
Acelerador
agarrado no fundo |
Genes Supressores
de Tumores: normalmente encontrados nas células que quando
danificados por mutações (recessivas) não mais codificam
proteínas que irão controlar os processos de divisão celular.
O mecanismo de controle de qualidade do ciclo celular se faz via indução
de apoptose. Quando ativados, levam à síntese de enzimas que irão
instruir a célula a cometer suicídio (apoptose). A eliminação
seletiva de células representa um meio adicional de controlar com precisão
o numero de células e suas atividades num tecido. Quando esses genes
estão anormalmente reprimidos, a sobrevivência prolongada
e aberrante facilitara o acumulo de mutações e ira contribuir
para a oncogênese.
|
Genes Supressores de Tumores
|
=
|
Freio |
|
Mutações nos Genes Supressores
de Tumores
|
=
|
Perda do Freio!
(Situação mais drástica!) |
Genes da Mobilidade Celular:
genes normalmente encontrados nas células, mas normalmente somente expressados
nas células moveis por natureza. Quando expressos nos oncócitos,
permite que estes produzam substancias (ex: Hialuronidase, TAF) que irão
dissolver as barreiras tissulares e permitir invasão de outros tecidos
e vasos sangüíneos. (INVASIBILIDADE).
Estes tipos de genes são fisiologicamente cooperativos,
e a seqüência de mutações varia de caso a caso, explicando
a imensa variação das historias clinicas dentro de um mesmo tumor.
A CASCATA DA CARCINOGÊNESE
VERSUS A QUEBRA DE VARIAS REPRESAS:
Mutações
em  |
Oncogene
|
Genes Supressores de Tumores e genes da apoptose
|
Genes da Mobilidade Celular
|
Acúmulo de mutações
|
Produzem

|
|
CÉLULA NORMAL
|
|
|
TRANSFORMAÇÃO MALÍGNA
|
CÉLULA PRÉ CANCEROSA
|
|
|
SELEÇÃO DE CLONES ATÍPICOS
|
ONCÓCITO - CÉLULA TRANSFORMADA
|
|
|
PROLIFERAÇÃO LOCAL
|
CARCINOMA IN SITU
|
|
|
INVASIBILIDADE |
CÂNCER
|
Observe que se ocorrer mutações nos genes supressores
de tumores, nos genes dos fatores de crescimento e nos genes da mobilidade celular,
mas não nos oncogenes, o Câncer não irá ocorrer até
que os oncogenes seja afetados. Nesse caso, quando os oncogenes forem ativados
por mutação, por que não haverá nenhum requerimento
a se cumprir para a completa cancerização, o tumor tenderá
a ocorrer de evolução extremamente rápida e letal, mesmo
sem apresentar a fase de carcinoma in situ.
Cada tipo de Câncer tende a ter um perfil particular
de lesões genéticas. Assim, a grande meta para os próximos
5-10 anos é caracterizar tais lesões genéticas para cada
tipo particular de câncer e assim estabelecer um catalogo de alterações
especificas e significativas para cada câncer especifico.
|
Gene
|
Tipo de tumor
|
| c-myc |
Carcinoma pulmonar de células
pequenas, Carcinoma mamário |
| N-myc |
Neuroblastoma, Carcinoma pulmonar
de células pequenas |
| L-myc |
Carcinoma pulmonar de células
pequenas |
| c-erbB |
Carcinoma pulmonar de células
pequenas |
| neu/erb-B2 |
Carcinoma mamário |
Uma vez que se disponha de tal catalogo, mesmo células
mostrando morfologia normal poderiam ser triadas para mostrar amplificações
de determinados genes e então ser determinado que células estão
em fase de Pré-cancer. Hoje em dia técnicas como Anticorpos monoclonais
anti proteína codificada por oncogenes, Hibridização in
situ, Southern Blot e Western Blot estão cada vez mais sendo usadas nessa
direção.
Um segundo objetivo e justificativa para a obtenção
de tal catalogo de lesões genéticas x câncer seria dar
subsídios para a terapia genética ("Gene Therapy"), na qual
se tentaria deletar o oncogene ou outros genes modificados por mutação
por técnicas moleculares ("Gene Knocking out" ou inserção
de genes invertidos /"anti-sense sequence").
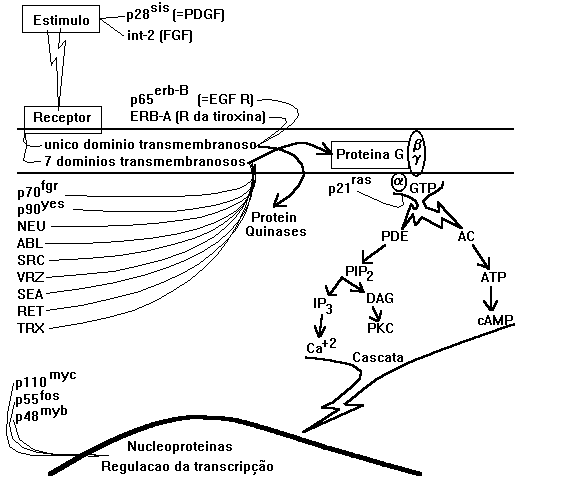
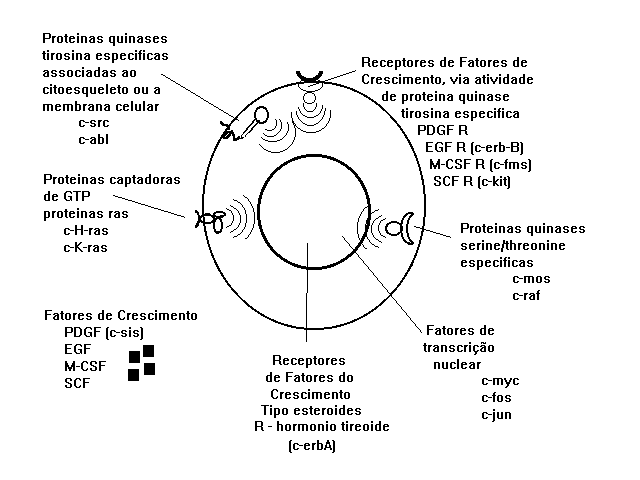
- Mecanismos
de ação dos oncogenes:
· Análogo
de um Fator de Crescimento secretado:
O p28sis mitógeno (Análogo ou subunidade ativa do PDGF) fosforila
o Fosfatidil-Inositol independentemente de Proteínas G. Ele é
codificado pelo gene do Simian Sarcoma Virus ("sis") .
· Homólogo
de Receptores de Fatores de Crescimento:
O ERB b é uma versão truncada do Receptor do EGF codificado
pelo gene do Avian Erythroblastosis Virus ("erb-b"). Esta forma do Receptor
não conta com o domínio de captação do EGF extracelular,
sendo ativo mesmo na ausência desse fator de crescimento.
O ERB a (p65erb a) é uma forma variante do receptor do hormônio
tireoideano
· Proteínas
Tirosina-Quinases:
50% de todos os oncogenes estão nessa categoria.
Autofosforilação com feed back Positivo, transformando o IP
em PIP2.
Rous Sarcoma Virus("src"); Abelson Murine leukemia Virus ("abl"), vrz, neu,
Yamaguchi 73 sarcoma Virus ("yes"), fgr
· Proteínas
Captadoras de GTP (Proteínas
G):
p21ras codificado pelo gene do Rat sarcoma Virus, mimetiza a proteína
G. Trata-se de uma unidade a com uma mutação
num ponto que modifica tão somente um aminoácido, (substituição
de glycina no códon 12) mas que com isso reduz ou mesmo impede a atividade
de GTPase, de maneira a impedir também a desativação
do sistema do Inositol. É como emperrar uma chave, de maneira a tornar
impossível o desligamento...
· Proteínas Reguladoras
da síntese de PIP2:
Proteínas que estimulam a síntese de Fosfatidil Inositol difosfato
(PIP2), conseqüentemente aumenta a síntese e a metabolização
do IP3, assim como a liberação de Ca+2.
Quando a síntese de PIP2 é bloqueada, ocorre redução
do Ca+2 intracitoplasmático.
Rous Sarcoma Virus ("src") e UR2 Sarcoma Virus ("ros") são
oncogenes que codificam proteínas com essa atividade.
· Proteínas
Reguladoras da Transcrição: Nucleoproteínas
(Fatores de transcrição)
que se conectam ao DNA participando diretamente na sua replicação.
p110myc , codificado pelo gene do Myelocytomatosis Virus; aumenta
a síntese de mRNA em 40 vezes, em 45 minutos.
p55fos, codificado pelo gene do FBJ osteossarcoma Virus; aumenta
a síntese de mRNA em 40 vezes, em 120 minutos.
p48myb, codificado pelo gene do Avian myeloblastosis Virus
Genes Supressores de tumores isolados
por Clonagem genética:
| Genes
Supressores de tumores |
Localização
Cromossômica |
Doença |
| APC |
5q21 |
Polipose Familiar |
| WT-1 |
11p13 |
Tumor de Wilms |
| Rb |
13q14 |
Retinoblastoma, Osteossarcoma |
| p53 |
17p12-13 |
Sindrome de Li-Fraumeni, vários
tumores |
| NF-1 |
17q11 |
Neurofibromatose de Von Recklinghausen |
| DCC |
18q21 |
Carcinoma do cólon |
- Fases da oncogênese
(ou Mecanismo de formação):
Com base na "Teoria da Co-Carcinogênese"
ou "do Crescimento Bifásico das neoplasias" ou ainda da "Iniciação
e Promoção" (paralelo com "Luz e revelador" em fotografia) temos:
- Iniciação
ou transformação maligna ou ainda cancerização
celular:
- Processo rápido ou lento, dependendo da potência
do "iniciador" (ou oncógeno ou carcinógeno). Modificação
irreversível e hereditária, que determina desdiferenciação
celular, ainda que desacompanhada de proliferação e invasividade.
A Exposição à carcinógenos (ppte substâncias
químicas, vírus, e radiações) ±
predisposição genética (proporção de
oncogenes e mutações acumuladas/ erros em transcrições
genéticas) ± aspectos multifatoriais
ligados ao ambiente (hábitos, dieta, atividades, idade, etc...)
= Mutações somáticas (modificações
no genoma) ou equivalente (desregulação gênica) determinando
a criação de clones celulares Atípicos com :
- Diminuição da velocidade do ciclo
mitótico (aumento da duração do ciclo celular);
- Aumento da fração de crescimento
(maior proporção de células em mitose)(maior nas
neoplasias indiferenciadas e nos imunodeprimidos);
- Aumento também do grau de perda celular
(conseqüência de "ataque imunológico", de compressão
pelas células adjacentes, de anormalidades bioquímicas
e metabólicas, de isquemias, de competição nutritiva,
de mitoses anormaisÞ Apoptose). O Maior
grau de perda celular não "e suficiente para compensar o Maior
Indice Mitotico.
- Latência:
- Seleção de clones atípicos que
apresentem:
- Vantagem de crescimento (maior viabilidade dos
oncócitos e maior ritmo de crescimento para vencer a concorrência
contra outros clones);
- Maior resistência a` vigilância imunológica
(através de secreções imunodepressoras, ou através
de "crescimento mudo"/ pequenas quantidades de neo-antígenos,
em vez de estimular síntese de anticorpos, pode inversamente
consumir os anticorpos que já existiam antes. Assim, com um crescimento
inicialmente muito lento, quando a neoplasia alcançasse volumes
necessários para realmente promover resposta imune, a neoplasia
já estaria volumosa o suficiente para não ser rejeitada.
- Após a seleção, ocorreria
a proliferação clonal que conduziria ao aparecimento de
brotos sólidos, respeitando ainda [na fase de latência]
a membrana basal.
- Promoção
ou proliferação neoplásica ou ainda cancerização
tissular:
- Decorre da ação continuada de substâncias
irritantes (denominadas "Co-carcinógenos" ou "Promotoras"), cujo
efeito não depende da dose ou potência e sim do tempo de
ação e da intensidade das reações que determinam.
A maior parte das substâncias envolvidas são iniciadoras
e promotoras, ainda que não simultaneamente (a atividade promotora
não ocorre paralelamente com a iniciadora). Por esta razão,
os promotores, além de induzir proliferação celular
e diminuir o período de latência, podem ainda diminuir a
dose necessária da substancia iniciadora quando a exposição
é simultânea.
- Trata-se de um processo lento, prolongado, caracterizado
por reação inflamatória hiperplásica e posteriormente
por autonomia proliferativa da célula transformada. Requer continuidade
e repetição. Determina aumento das discarioses (as alterações
cromossômicas aumentam com a evolução das neoplasias)
e também das cariocineses (com elevação do índice
mitótico e aumento da probabilidade de mutações),
e conseqüente aumento da população de oncócitos
pelo aumento da "fração de crescimento" (aumenta o número
de células em mitose, ainda que diminua a velocidade do ciclo mitótico).
Assim é a fase de promoção que leva à exteriorização
ou expressão clínica da neoplasia.
· A proliferação
oncocitária teria um "Tempo
de duplicação" (Intervalo de tempo que ele ocupa para
atingir o dôbro de seu volume inicial) que variaria entre 8 a 600 dias
(mais freqüentemente entre 20 a 100 dias), e tenderia a aumentar a medida
que a neoplasia "envelhece". Isto caracterizaria um crescimento exponencial
decrescente, também chamado de "Crescimento Gompertziano". O aumento
no tempo de duplicação decorreria do aumento da população
de células em diferenciação (fora de mitoses) determinando
diminuição da fração de crescimento. A importância
disso reside no fato de que sabendo-se o volume da neoplasia, poderia-se estimar
o número de células que a comporia, e desta maneira se ter uma
idéia do número de "Tempo de duplicações" já
ocorridos. A história natural das neoplasias malignas dura em geral
40 a 45 T.D.s, o que corresponde à presença de aproximadamente
1 trilhão (1012) de oncócitos. Esta enorme população
incoordenada geralmente é incompatível com a vida. As neoplasias
menores e já detectáveis clinica e macroscopicamente possuem
1 cm de diâmetro, correspondendo a aproximadamente 1 bilhão de
oncócitos, com ± uns 30 T.D.s ( correspondendo
a uns 2/3 do crescimento total potencial estimado para todo tumor).
· Com o aumento das
cariocineses e das discarioses nos clones atípicos se acentuaria a
perda de inibição por contato. Posteriormente, com a secreção
do TAF ("Tumor Angiogenesis Factor") haveria um crescimento da vascularização
dentro da massa neoplásica, aumentando dramaticamente o ritmo de crescimento
e a invasividade desta.
- Conceito:
Grupo de lesões, geralmente epiteliais, que são potencialmente
cancerosas, ainda que não necessariamente e invariavelmente evoluam
para o câncer. No entanto, com uma freqüência significativa
precedem as neoplasias.
Algumas controvérsias:
- Leucoplasia x atipia celular ?
- Enquadramento de coristias, hamartias, e heterotopias
(sem suporte clínico, estatístico e experimental);
- "Displasia" mamaria" [hiperplasia/ mastopatia fibrocística]
(respostas diferentes e independentes às disendocrinias).
- Nomenclatura
e classificação:
- Displasia (gr."dis"
= dificuldade/ distúrbio +"plásso" = formar): Distúrbio
na formação. Conceito muito abrangente e confuso.
- Termo genérico que, afetando um órgão,
abrange vários distúrbios do desenvolvimento e do crescimento
(processos regressivos, freqüentemente ligados a` condições
genéticas). Exemplos: displasia Coxo-Femural, displasia fibrosa
do osso, etc...
- Quando afeta um tecido, abrange os erros locais
de desenvolvimento (Hamartias/omas e Coristias/omas)
e as "displasias adquiridas" ou "Hiperplasias atípicas", que
são as verdadeiras lesões pré-malignas e afetam
principalmente as mamas e cérvix. Trata-se de uma resposta proliferativa,
comum nas irritações crônicas, acompanhadas de perda
de diferenciação, de atipia celular (pleomorfismo e hipercromasia)
e perda da progressão regular normal de células germinais
a` células totalmente diferenciadas, com mitoses em posições
anormais.
- "Carcinoma in situ" ou "Ca
pré invasivo": É uma hiperplasia atípica com
todas as características de neoplasia maligna, exceto a capacidade
invasiva. Está confinado a` camada epitelial, provavelmente devido
ao fato de que ainda não ocorreu proliferação vascular
na massa neoplásica (SLAUSON & COOPER, 1984).
- Leucoplasia: Espessamento
hiperplásico circunscrito, esbranquiçado, do epitélio
pavimentoso das mucosas [oral, labial, esofágica, vesical, cérvico-uterina,
vulvar, etc...], com acantose, hiperceratose, e as vezes atipias celulares.
- Outras lesões pré malignas (pouco interesse
em medicina veterinária.): Mastopatia fibrocística,
atipias epiteliais da cérvix uterina, ceratose actínica
senil, "Doença de BOWEN", "Eritroplasia de QUEIRAT", "Doença
de PAGET", xeroderma pigmentoso, radiodermites, adenomas polipoides do
cólon e estômago, metaplasia intestinal da mucosa gástrica,
polipose vesical, nevos pigmentados salientes, etc...
- GRADUAÇÃO E
ESTADIAMENTO DE UMA NEOPLASIA:
- A malignidade
de uma neoplasia varia conforme o tipo e a fase (inicial,
avançada ou terminal) da mesma. A graduação
da malignidade pode ser feita microscopicamente considerando o grau
de diferenciação dos oncócitos, a invasividade e o
índice mitótico da neoplasia.
- O estadiamento
(ou estagiamento) é mais uma forma de avaliação
clínica que anatomopatológica da evolução dos
tumores. Leva em consideração várias técnicas
como Raio X, Laparoscopia, biópsias, tomografia, ultra-sonografia
e exames de patologia clinica (detecção dos chamados "Marcadores
Tumorais"- ex: "Proteína de Bence-Jones" no Plasmocitoma/Mieloma
Múltiplo; "alfa proteína fetal" no Carcinoma Hepatocelular
e Teratoma testicular; "Antígeno carcinoembrionário"/CEA nas
neoplasias do trato respiratório, gastrointestinal, pâncreas
e mama; "Fosfatase Alcalina" em várias neoplasias).
- O método de estadiamento mais utilizado em medicina
para as neoplasias em geral é o "TNM" da União Internacional
Contra o Câncer, publicado em 1958. "T" significa as qualidades do
Tumor primário (diâmetro da massa, invasão e
infiltração nos tecidos vizinhos). "N" significa linfoNodos
e indica o comprometimento ou não dos linfonodos regionais. "M" significa
Metástases e realça sua presença. A principal
falha dos sistemas de estagiamento é a sua incapacidade de abranger
os numerosos casos de neoplasia aparentemente local ou regional onde estão
presentes micrometástases indetectáveis aos métodos
diagnósticos atuais.

· Bases:
- Oncologia experimental (ppte em camundongos atríquicos
e carcinógenos químicos),
- Patologia humana e animal espontânea,
- Cancerização "in vitro" (as culturas
de tecidos, após "n" gerações, geralmente sofrem transformação
maligna).
· Teorias
da oncogênese:
- Da irritação crônica (VIRCHOW);
- Do desvio embrionário (CONHEIM);
- Da diferenciação defeituosa pós
embrionária (RIBERT)
- Do desvio metabólico (WARBURG, 1925/25):
"O crescimento descontrolado dos oncócitos se relaciona com desvio
metabólico expresso pela glicólise aeróbica".
- Da uniformização bioquímica
dos oncócitos (GREENSTEIN, 1955)
- Da deleção de repressores (MILLER,
1945): Perda ou alteração permanente de fatores protéicos
capazes de controlar o crescimento celular.
- Da mutação (de células somáticas,
por cancerígenos):
* Teoria da mutação
(de células somáticas, por cancerígenos):
· .
Argumentos favoráveis:
- As alterações celulares são hereditárias
(i.e., as características dos oncócitos são transmitidas
a` descendência);
- O UltraVioleta é mutagênico (dímeros
de thymidine no DNA) e também oncogênico.
- A grande maioria das neoplasias naturais são
monoclonais.
- São necessárias "n" alterações
no DNA para se originar um câncer, e se um indivíduo já
é portador de alguma, existe maior predisposição
("Multiple hit hipothesis").
- A inibição no reparo ao DNA pós
replicação diminui a carcinogênese, mas aumenta a
toxidez do carcinogênico.
- No xeroderma pigmentoso, existe maior capacidade de
reparo às lesões no DNA, aumentando assim a freqüência
de reparos pós replicação e a probabilidade de erros
na reparação predispondo às mutações
e a` oncogênese.
Mecanismos
celulares de reparo ao DNA:
Excisional (remoção e substituição
do segmento lesado. É livre de erro).
Pós replicação (em lesões
extensivas, com alta probabilidade de erros/mutação. As
fitas lesadas são replicadas em suas regiões íntegras,
com fragmentos resultantes recombinando-se).
- Argumentos desfavoráveis:
- Os hidrocarbonetos policíclicos são
oncógenos poderosos, mas mutágenos escassos e "n" mutágenos
fortes não são carcinógenos.
* Teoria da perda
dos mecanismos de controle (defeitos nos mecanismos epigenéticos):
*Teoria do Mêmbron
(PITOT, 1969):
· A deleção
enzimática derivaria de alterações no mosaico das unidades
de membrana do RE, diminuindo a estabilidade da associação
RNAm-Sítio de membrana (mêmbrom), descontrolando assim a síntese
de proteínas específicas.
* Teoria Viral:
· Generalizante
demais. Na maior parte das neoplasias Não pode ser demonstrada a
existência de vírus (argumento desfavorável), talvez
pelo "mascaramento" ou "lisogenização" (integração
total ou parcial do genoma vírico no genoma celular gerando uma mutação/
inserção do vírus através de ligações
com radicais livres determinando modificações na sucessão
das bases do DNA (HASUMI, 1986 - aponta 84 tipos de vírus oncogênicos).
* Teoria do Próvirus
:
· Células
susceptíveis a` carcinogênese possuem estruturas lábeis
no DNA, capazes, sob estímulo adequado, de se tornarem livres e independentes.
Os segmentos livres ("Pró- vírus"), seriam capazes de replicação
e de integração noutros pontos de seqüência do
DNA.
* Teoria do virogene
ou protovirus (BALTIMORE,
1970; TEMIN, 1970/71):
· A
seqüência de DNA ("Protovirus") de uma célula X codifica
um RNA que, agindo como um vírus, se transfere para outra célula(Y),
e codifica nesta um DNA idêntico ao protovirus, que se integra no
DNA de Y provocando mutação. A transcrição do
virogene ou do protovirus em RNA e sua tradução em proteínas
pode ocorrer com intensidade e extensão variáveis, dependendo
da regulação e interação de genes. A expressão
parcial do virogene determinaria a transformação maligna,
enquanto a expressão total do virogene inteiro levaria a` produção
de novos vírions.
· A questão
que fica é quem veio primeiro? (o ôvo ou a galinha?/ os oncogenes
são de origem celular e se incorporaram a alguns vírus/"Protovirus",
ou se originaram dos vírus e foram por eles incorporados nas células/"virogene"
?).
* Teoria dos oncogenes
(CROCE, 1977; STRONG, 1977; PHILLIPS-QUAGLIATTA,
1978; ARRIGHI, 1980);
· A
causa primária das neoplasias seria uma desregulação
gênica, i.e. uma alteração no sistema genético
e epigenético complexo responsável pela regulação
da reprodução e diferenciação celular. De acordo
com esta teoria, todas as células teriam uma bateria de genes (reguladores
e estruturais). Nesta bateria haveriam genes do crescimento, cuja função
seria determinar e regular a reprodução e a diferenciação
celular. Tais genes, por mutação, originariam os "oncogenes",
variáveis e hereditários (donde a predisposição
familiar às neoplasias e lesões pré malignas). Os oncogenes
ocasionariam transformação maligna quando:
- a mutação afetasse a expressão
dos genes do crescimento;
- a mutação afetasse os genes reguladores
dos genes do crescimento. A deficiência no sistema de regulação
poderia ser o causador de desrepressão e/ou ativação
do crescimento celular.
- Esta deficiência no sistema de regulação
poderia ser por:
- Alteração nos mecanismos de reparo
do DNA, favorecendo o acúmulo de mutações; ou
- Alteração nos mecanismos epigenéticos
(nos processo normais de regulação gênica, envolvendo
mais diretamente proteínas e RNA e não o DNA).
- a mutação decorresse da introdução
de oncogenes exógenos desconhecidos dos mecanismos de regulação
gênica, veiculados pelos vírus oncogênicos (Obs:
Nem todos vírus oncogênicos portam oncogenes, alguns
são meramente mutagênicos, quando se inserem nas seqüências
dos genes reguladores do crescimento).
Sabe-se hoje que a expressão dos oncogenes
é também detectável em células normais,
em baixo grau, estando amplificada nos oncócitos. Esta observação
sugere um "efeito de dosagem" (tipo "cascata"/ como a ativação
do complemento e da coagulação do sangue; ou tipo "rede
auto-reguladora"/ como no sistema imunológico) como responsável
pela transformação maligna.
 De volta ao Laboratório de Apoptose
De volta à Patologia Geral
De volta ao Laboratório de Apoptose
De volta à Patologia Geral

 Para
referenciar o material desta publicação: Anilton Cesar
Vasconcelos. Patologia Geral em Hipertexto. Universidade Federal de
Minas Gerais. Belo Horizonte, Minas Gerais, 2000. Se você tiver alguma
sugestão ou comentários, por favor envie-os para: Laboratório
de Apoptose/ Departamento de Patologia Geral/ Instituto de Ciências
Biológicas da Universidade Federal de Minas Gerais, Belo Horizonte,
MG 31270 010
Para
referenciar o material desta publicação: Anilton Cesar
Vasconcelos. Patologia Geral em Hipertexto. Universidade Federal de
Minas Gerais. Belo Horizonte, Minas Gerais, 2000. Se você tiver alguma
sugestão ou comentários, por favor envie-os para: Laboratório
de Apoptose/ Departamento de Patologia Geral/ Instituto de Ciências
Biológicas da Universidade Federal de Minas Gerais, Belo Horizonte,
MG 31270 010
