Deposição, Infiltração ou "Degeneração"
Amiloide.
Conceito:
Deposição intersticial de material fibrilar glicoproteico,
de composição química variável, de aspecto hialino
(ainda que tintorialmente diferente das hialinoses), eosinofílico,
amorfo, homogêneo, birrefringente, metacromático, especialmente
nas membranas basais, com reações tintoriais específicas.
Histórico:
Rudolf Virchow, ao verificar reação de metacromasia do
Iodo (de amarronzado para azulado, semelhantemente ao que ocorre com amido),
batizou de amiloide, acreditando ser o material depositado de natureza glicílica.
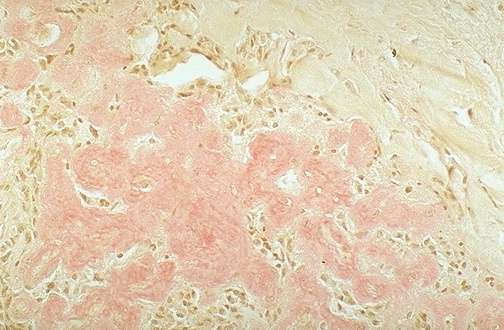
Características Macroscópicas:
- Aumento de volume e de consistência;
- Palidez (isquemia pela compressão
vascular, associada à própria natureza da substância
amiloide);
- Tensão capsular e friabilidade
(predisposição à rupturas/ não se recomenda
biópsias hepáticas quando se suspeita de amiloidose. Tal procedimento
poderia causar rupturas e hemorragias graves pela diminuição
dos fatores de coagulação);
- Superfície de corte pálida,
translúcida, homogênea, (lardácea tipo toucinho
quando a deposição amiloide é difusa) ou
"em Sagú" (se focal, perifolicular);
- Reação de metacromasia característica
com o Iodo.
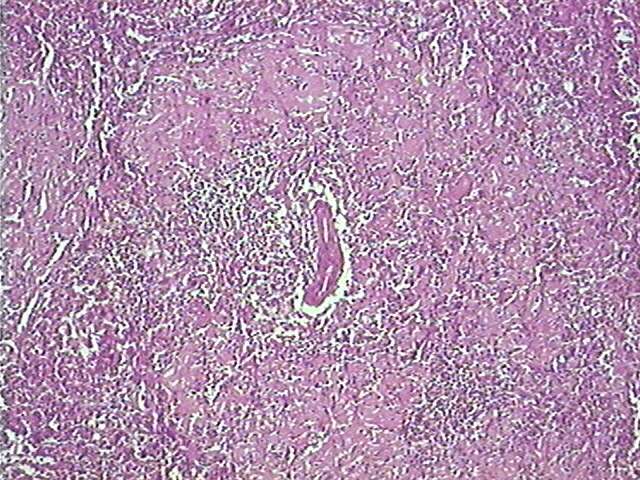
Características Microscópicas:
Material hialino, eosinofílico, amorfo, homogêneo, birrefringente,
metacromático (i.e., cora-se com cor diferente daquela que cora
os demais tecidos);
Histoquímica: Róseo à
HE; PAS positivo; Vermelho (em vez de verde ou roxo) aos corantes Verde
Metileno, Violeta de Cresila e Violeta de Genciana. O melhor corante
para a identificação do amiloide é o Vermelho Congo
(que cora o amiloide em alaranjado
em contraste com o róseo de fundo). Ao exame com luz polarizada,
o amiloide corado com Vermelho Congo mostra uma refringência verde
(as vezes vermelha ou azul amarelada) e ao exame com Ultra Violeta mostra
uma fluorescência avermelhada.
Sítios de predileção:
Glomérulos renais, sinusóides hepáticos, e em volta
dos folículos esplênicos, sempre diminuindo o espaço
e comprimindo as células.
Composição
Química:
Variável - num mesmo indivíduo, entre indivíduos
e entre espécies;
Não existe "uma" substância Amilóide, mas sim várias
(AL - oriunda de cadeia leve de Ig´s
ou parte da cadeia k ou l,
AA
- proteína de fase aguda, sintetisada no fígado, após
estímulo de IL-1, IL-6 e/ou TNF-a,
b2
m ou microglobulina b2
- componente do MHC I, presente no soro normal, Ab2
ou Proteína amilóide b2 - derivada da proteína precursora
APP, AE - de origem endócrina,
etc) . São, no entanto, sempre polipeptídeos dispostos
em arranjo molecular especial, constatável à Microscopia
eletrônica de transmissão. Assim, o termo Amilóide
define uma estrutura molecular física e não uma composição
química;
A substância Amilóide é composta de:
- Proteínas:
Principalmente com aminoácidos acidificantes. Não
há Hidroxilisina nem hidroxiprolina como no colágeno, na
elastina ou na fibrina. Os aminoácidos acidificantes (em inúmeras
seqüências diferentes) formam peptídeos empilhados e
dobrados ("pregueamento Beta" - que lhe confere característica
resistência à degradação) formando uma estrutura
molecular em lâmina ou folha pregueada antiparalela, semelhante
à da Beta ceratina e à da seda, quando observada com
difração de raios X.
- 5 a 10% de Carbohidratos: principalmente
galactose, glicose e manose. Explica a reação com PAS e
com o Iodo.
- 10% de lípides: principalmente
colesterol, esteres do colesterol, triglicerídeos, ácidos
graxos e fosfolípides.
O Amilóide é formado por fibrilas finas e de comprimento
variável (70 a 100 Ao de diâmetro por 100 a 10.000 Ao de
comprimento), não ramificadas, discretamente encurvadas, dispostas
irregularmente e embebidas em matriz homogênea.
Origem:
Parece estar ligada à fagocitose de imunocomplexos e proteínas
desnaturadas circulantes pelos fagócitos sangüíneos ou
do Sistema Mononuclear Fagocitário (ex. SRE), com degradação
enzimática (cisão) de várias proteínas e polipeptídeos
e exocitose de cadeias leves de globulinas, precursoras do Amilóide.
Estes corpos residuais exocitados é que então forneceriam
o substrato para a polimerização das fibrilas.
Classificação(Tipos
etiopatogenéticos de Amiloidose):
- Amiloidose de origem imunológica:
Causada em última análise por alguma disfunção
do Sistema Imunológico. Existem 2 tipos:
- Amiloidose Primária:
Decorre de um descontrole da resposta imunológica/ "Discrasia
de Plasmócitos". É vista no Plamocitoma ou Mieloma
Múltiplo, quando plasmócitos anormais aumentam a produção
de precursores de globulinas e de Amilóide tipo B (de origem
imunoglobulínica). Os sítios de deposição
mais freqüentes são o coração, os pulmões,
os músculos, a língua, os pequenos vasos, a pele, o trato
gastrointestinal e os nervos.
- Amiloidose Secundária:
Decorre da exaustão do Sistema Imunológico, em
conseqüência de superestimulação e/ou da
carência de fatores que o tornem eficiente. É visto
nas Doenças Crônicas Caquetizantes (Tbc, Osteomielite,
lepra, sífilis, artrite reumatóide, carcinomas e sarcomas,
etc...) e em animais utilizados na produção de soro
hiperimune, quando o Amilóide que predomina é o do
tipo A e C (de origem desconhecida). Os sítios de deposição
mais freqüentes são o fígado, o baço, os
rins, a adrenal, o pâncreas e os linfonodos. O mecanismo mais
provável é o da supressão da ação
dos linfócitos T com manutenção da imunidade humoral
ou o do esgotamento do mecanismo pelo qual imunocomplexos ou proteínas
desnaturadas são normalmente eliminadas da circulação
pelo Sistema Monocítico Fagocitário. Com a catabolização
incompleta, resíduos são depositados nos tecidos.
- Amiloidose de origem endócrina:
Deposição de Amilóide em concomitância à
ocorrência de neoplasias de origem neuroectodérmica
("Amiloide Apud" - devido às características das células
secretoras de hormônios polipeptídeos com as quais está
associada). É vista no Carcinoma medular da tireóide, no
gastrinoma, no feocromocitoma e no insulinoma - quando se formam complexos
de precursores de hormônios polipeptídeos análogos às
subunidades de Amilóide.
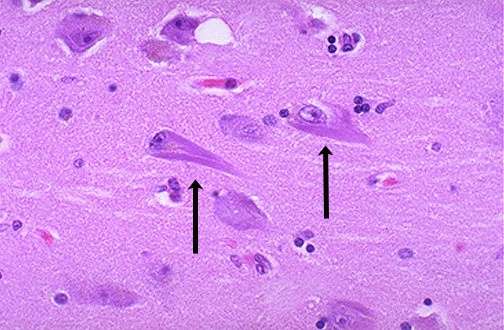
- Amiloidose localizada, de origem neurológica:
Doença de Alzheimer,
Scrapie, e encefalopatias espongiformes (Doença da Vaca
Louca, Doença de Creutzfeldt-Jakob, Síndrome de
Gerstmann-Straussler-Scheinker, etc...).
Conseqüências:
- A deposição progressiva de Amilóide acaba levando
à atrofia compressiva do parênquima
adjacente.
- Nos rins: É um dos componentes da "Síndrome
Nefrótica" (Proteinúria + hipertensão arterial
+ Insuficiência renal com uremia + obliteração glomerular
com diminuição da filtração glomerular).
- No coração: Leva à "Insuficiência
Cardíaca refrataria aos digitálicos".
- Na adrenal: Pode determinar "Doença
de Addison" (Insuficiência adrenal).
- No fígado e baço: Compressão parenquimatosa +
friabilidade = Predisposição à
rupturas (que no caso do fígado geralmente determina hemorragia
grave e letal em virtude da diminuição dos fatores da coagulação).


{kind=link}
{kind=link}
{kind=link}